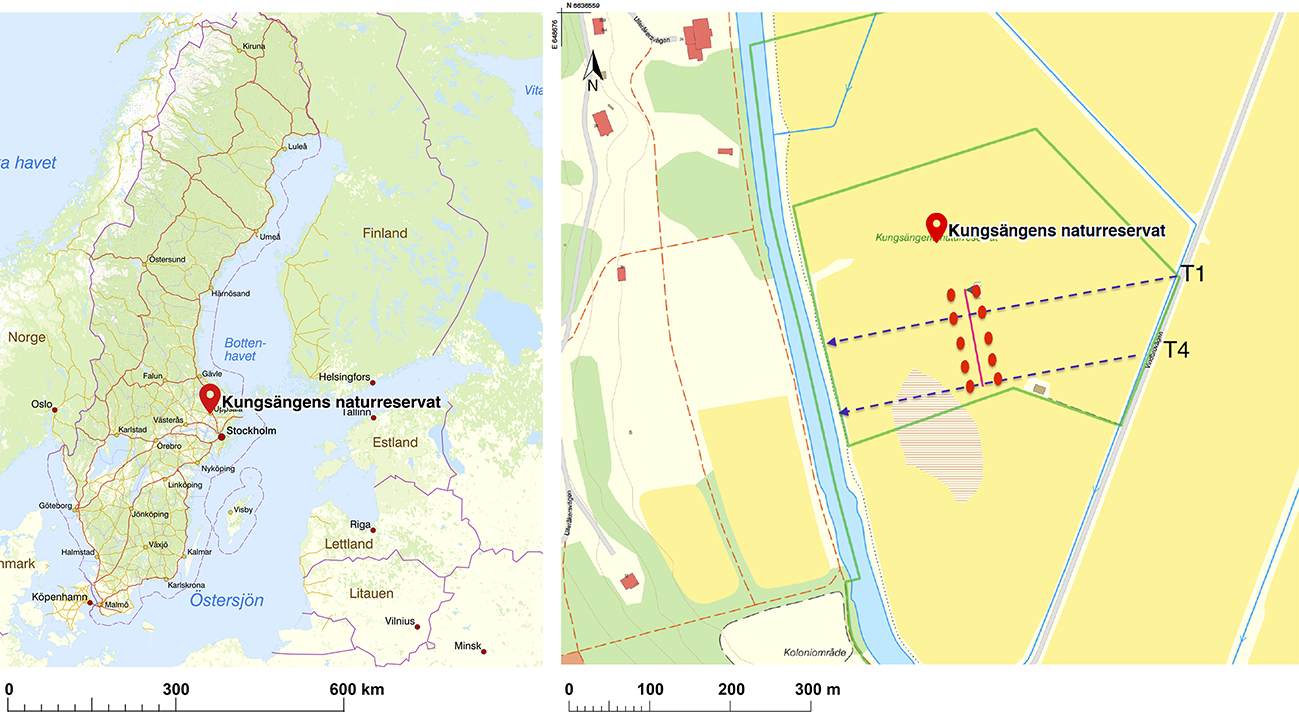
Fungi form diverse communities and play essential roles in many terrestrial ecosystems, yet there are methodological challenges in taxonomic and phylogenetic placement of fungi from environmental sequences. To address such challenges we investigated spatio-temporal structure of a fungal community using soil metabarcoding with four different sequencing strategies: short amplicon sequencing of the ITS2 region (300–400\ bp) with Illumina MiSeq, Ion Torrent Ion S5, and PacBio RS II, all from the same PCR library, as well as long amplicon sequencing of the full ITS and partial LSU regions (1200–1600\ bp) with PacBio RS II. Resulting community structure and diversity depended more on statistical method than sequencing technology. The use of long-amplicon sequencing enables construction of a phylogenetic tree from metabarcoding reads, which facilitates taxonomic identification of sequences. However, long reads present issues for denoising algorithms in diverse communities. We present a solution that splits the reads into shorter homologous regions prior to denoising, and then reconstructs the full denoised reads. In the choice between short and long amplicons, we suggest a hybrid approach using short amplicons for sampling breadth and depth, and long amplicons to characterize the local species pool for improved identification and phylogenetic analyses.