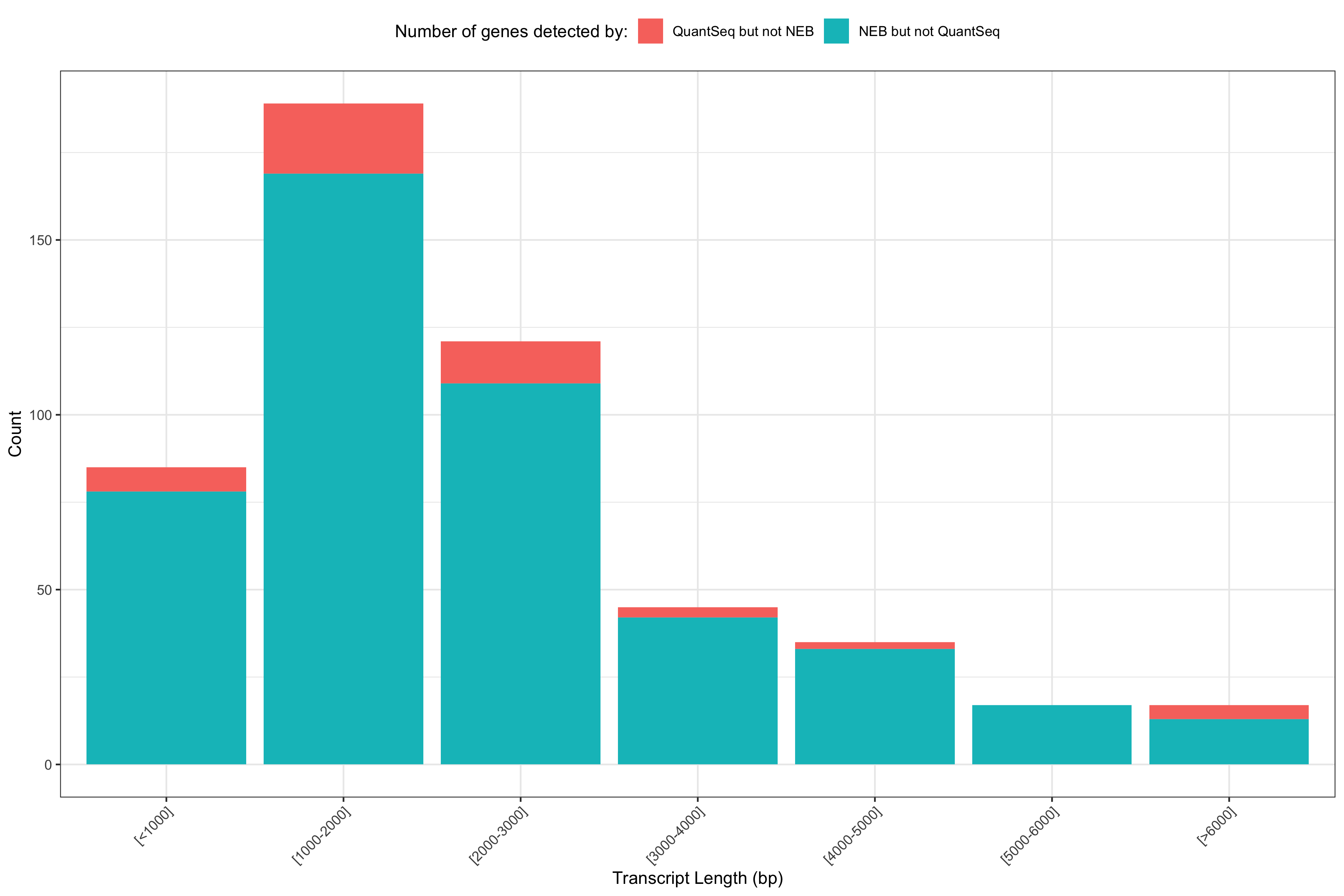
RNA sequencing (RNA-Seq) is a popular method for measuring gene expression in non-model organisms, including wild populations. While RNA-Seq can measure gene expression variation among wild-caught individuals and can yield important biological insights into organism function, sampling methods may also influence gene expression estimates. We examined the influence of multiple technical variables on estimated gene expression in a non-model fish, the westslope cutthroat trout (Oncorhynchus clarkii lewisi), using two RNA-Seq library types: 3’ RNA-Seq and whole mRNA-Seq. We evaluated effects of dip netting versus electrofishing, and of harvesting tissue immediately versus 5 minutes after euthanasia on estimated gene expression in blood, gill, and muscle. We detected 30% more genes with whole mRNA-Seq than with 3’ RNA-Seq and found that 58% of genes were significantly differently expressed between 3’ RNA-Seq and whole mRNA-Seq. Our findings indicate that 3’ RNA-Seq and whole mRNA-Seq are robust to the technical variables related to the field sampling approaches tested here with a lack of differential gene expression among sampling methods and tissue collection time after euthanasia. However, we found that gene expression varied based on which RNA-Seq library type was used on the same set of samples. Our study suggests researchers could safely rely on different fish sampling strategies in the field and save money and analyze more individuals using 3’ RNA-Seq, but should use whole mRNA-Seq when working with a species without good genomic resources, and when maximizing the number of genes identified and detecting alternative splicing are important.