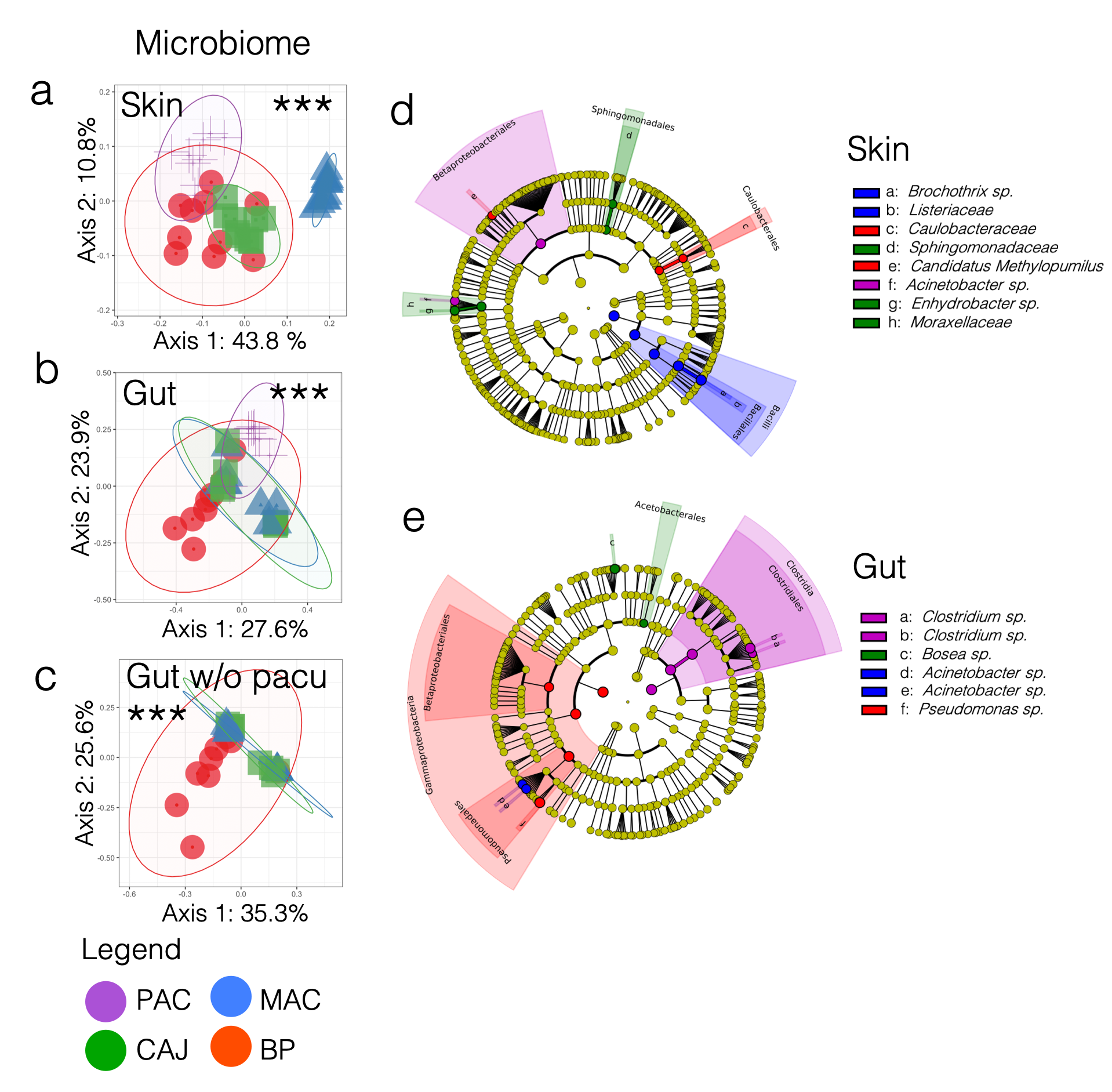
The study of the eukaryotic fraction of the microbiota using a metabarcoding approach is usually hindered by the high host to eukaryotic microbiota DNA ratio in samples. Indeed, the 18S rRNA gene is very similar for both the host and its eukaryotic communities, leading to a preferential amplification of the predominant host DNA when using universal primers. Multiple approaches have been developed to reduce host DNA amplification. One method is based on elongation arrest blocking primers, oligonucleotides modified with a C3 Spacer that stops the advancement of the DNA polymerase at non-conserved regions of a target gene. In this paper, we successfully developed and tested species-specific elongation arrest blocking primers to block the Flag cichlid, Mesonauta festivus, 18S rRNA SSU. Our elongation arrest blocking primers significantly reduced the amount of host DNA amplicons by 66 %. In addition to reducing the amount of sequencing wasted, the blocking primers increased the detectability of potentially dangerous parasitic taxa in fish gut, highlighting the potential of the method for parasitic screening. For instance, we discovered a case of infection by the parasitic ciliate Nyctotherus sp. and detected the presence of a parasitic Trematode and an Amoebae. While our data support the possibility of achieving a complete inhibition of host DNA amplification using elongation arrest blocking primers, more research is still required. Still, there is a need for the development and additional testing of protocols to study the eukaryotic diversity present in fish gut, a slow-growing field of study in comparison to its prokaryotic counterpart.