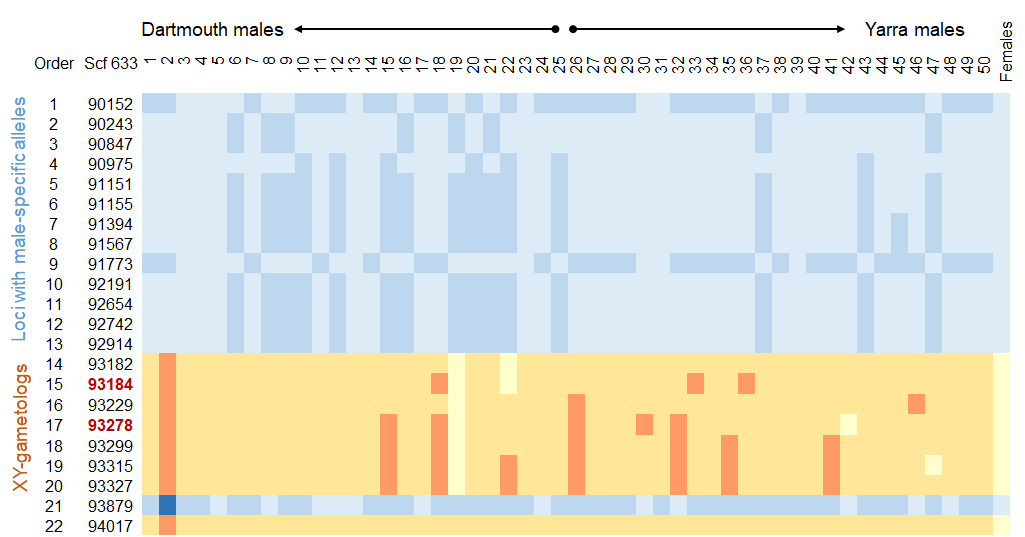
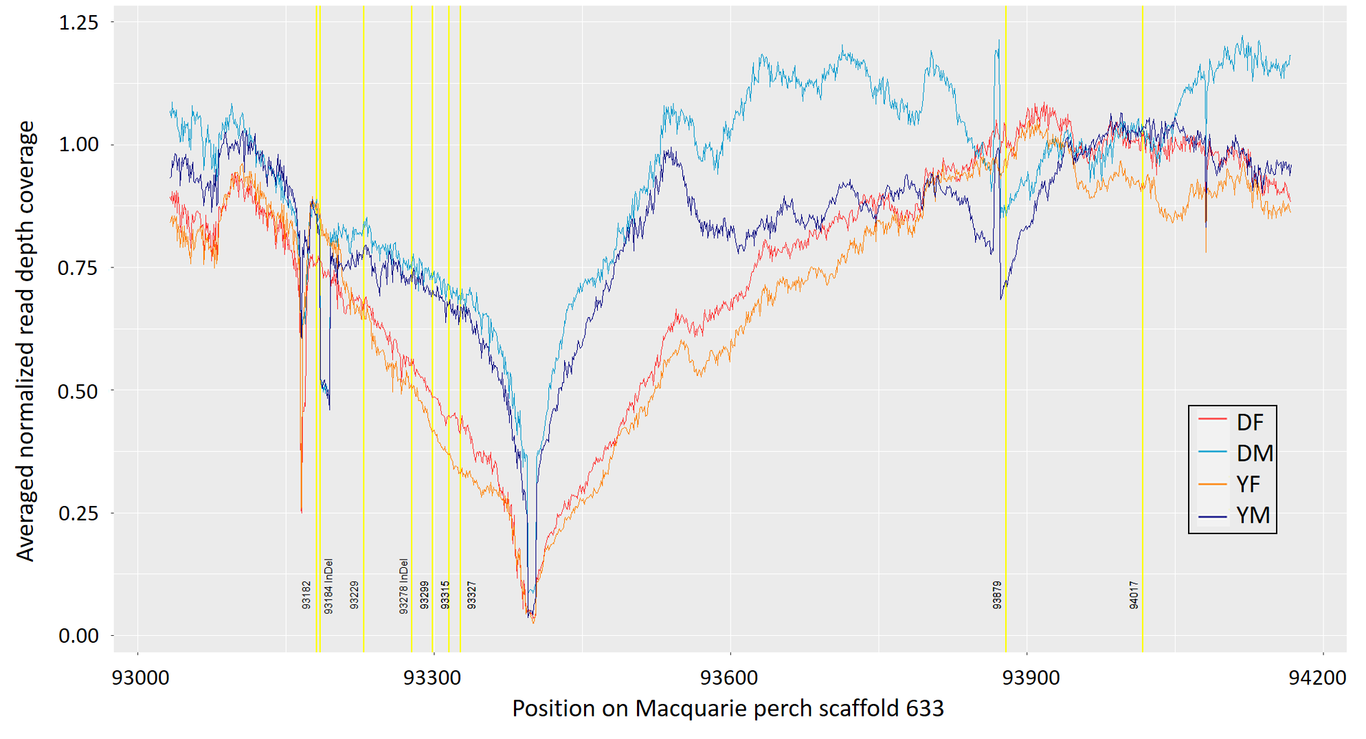
Sex-specific ecology has management implications, but rapid sex-chromosome turnover in fishes hinders development of markers to sex monomorphic species. Here, we use annotated genomes and reduced-representation sequencing data for two Australian percichthyids, the Macquarie perch Macquaria australasica and the golden perch M. ambigua, and whole genome resequencing data for 50 Macquarie perch of each sex, to detect sex-linked loci, identify a candidate sex-determining gene and develop an affordable sexing assay. In-silico pool-seq tests of 1,492,004 Macquarie perch SNP loci revealed that a 275-Kb scaffold, containing the transcription factor SOX1b gene, was enriched for gametologous loci. Within this scaffold, 22 loci were sex-linked in a predominantly XY system, with females being homozygous at all 22, and males being heterozygous at two or more. Seven XY-gametologous loci were within a 146-bp region. Being ~38 Kb upstream of SOX1b, it might act as an enhancer controlling SOX1b transcription in the bipotential gonad that drives gonad differentiation. A PCR-RFLP sexing assay, targeting one of the Y-linked SNPs, tested in 66 known-sex Macquarie perch and two individuals of each sex of three confamilial species, and amplicon sequencing of 400 bp encompassing the 146-bp region, revealed that the few sex-linked positions differ between species and between Macquarie perch populations. This indicates sex-chromosome lability in Percichthyidae, also supported by non-homologous scaffolds containing sex-linked loci for Macquarie- and golden perches. The resources developed here will facilitate genomic research in Percichthyidae. Sex-linked markers will be useful for determining genetic sex in some populations and studying sex chromosome turnover.