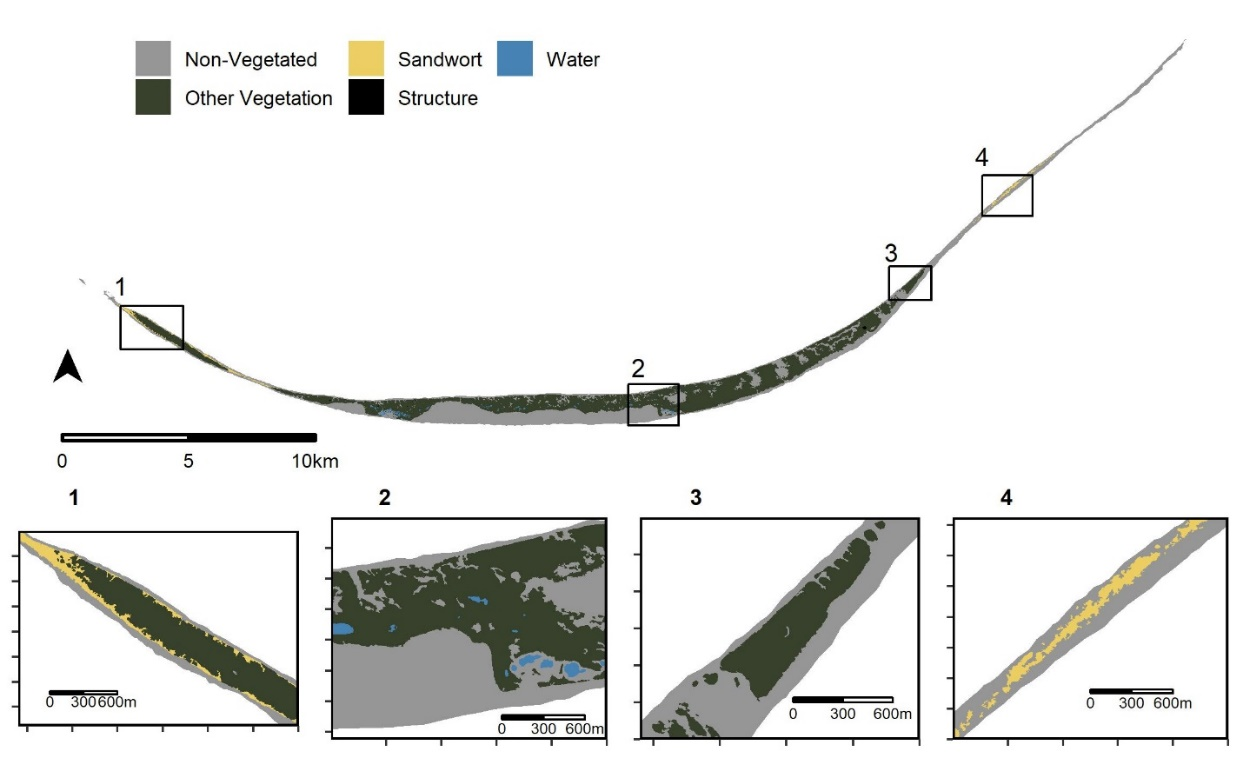
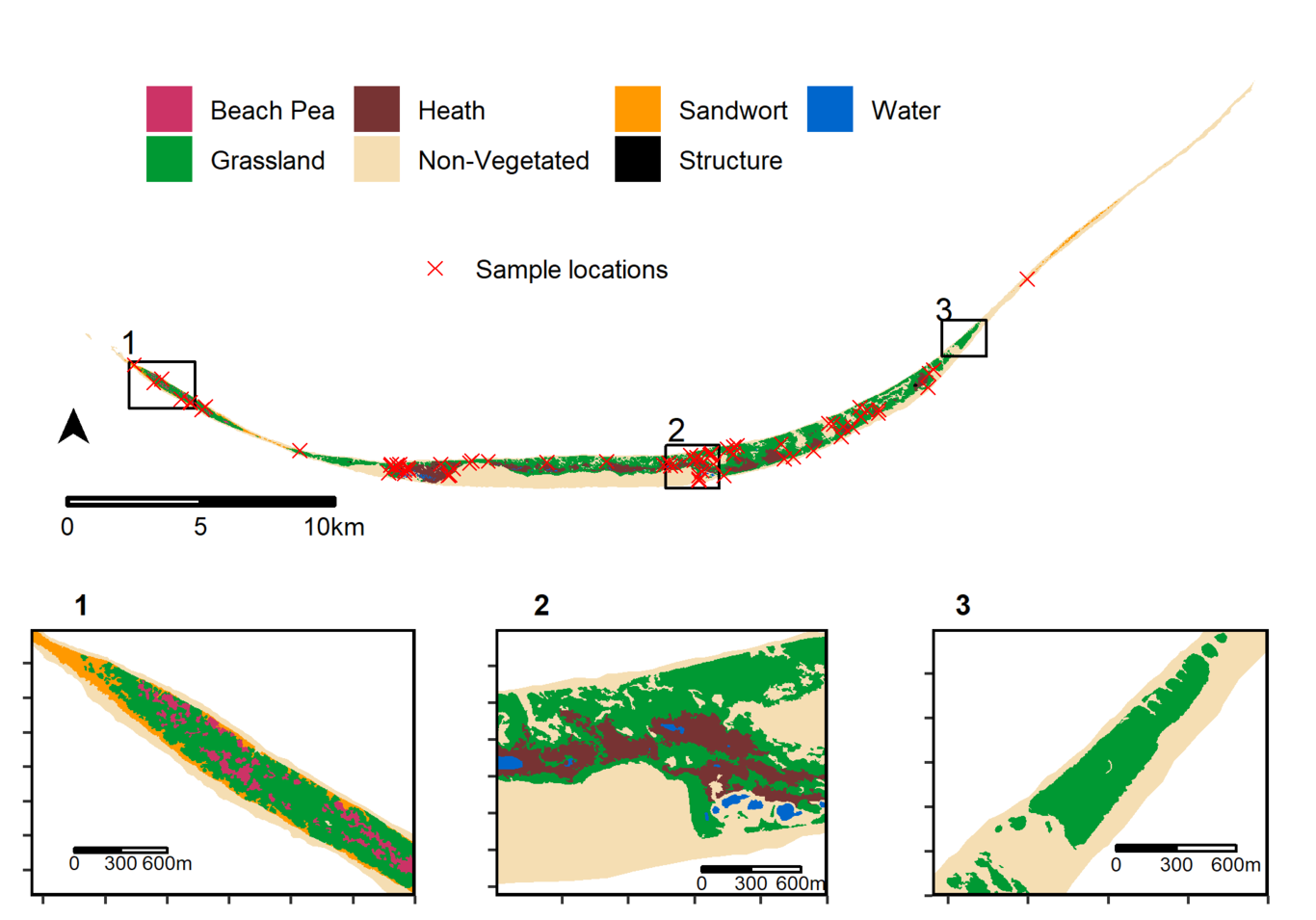
Studies of microbiome variation in the wild often emphasize host physiology and diet as proximate selective pressures acting on host-associated microbiota. In contrast, microbial dispersal is more rarely considered, and when it is, spatially autocorrelated environmental variables are sometimes overlooked. Using amplicon sequencing, we characterized the bacterial microbiome of adult female (n = 86) Sable Island horses (Nova Scotia, Canada) as part of a detailed, individual-based study of the ecology and evolution of this unmanaged free-living population. Using data on sampling date, horse location, age, parental status, and local exposure to habitat variables, we contrasted the ability of spatiotemporal, physiological, and environmental factors to explain microbiome diversity among Sable Island horses. We extended inferences made from these analyses with both phylogeny-informed and phylogeny-independent null modeling approaches to identify deviations from stochastic expectations. Phylogeny-informed diversity measures were more often correlated with local habitat composition, although null modeling results did not support differential selection acting on the microbiome as the mechanism for these correlative patterns. Conversely, phylogeny-independent diversity measures were best explained by spatial terms, with evidence for spatial- and host social-structured bacterial dispersal limitation. Parental status was important but correlated with measures of β-dispersion rather than β-diversity (mares without foals had lower alpha diversity and more variable microbiomes than mares with foals). Our results suggest that inter-host microbiome variation in this population is driven more strongly by bacterial dispersal limitation and ecological drift than by differential selective pressures, highlighting the need to consider alternative ecological processes in the study of microbiomes.