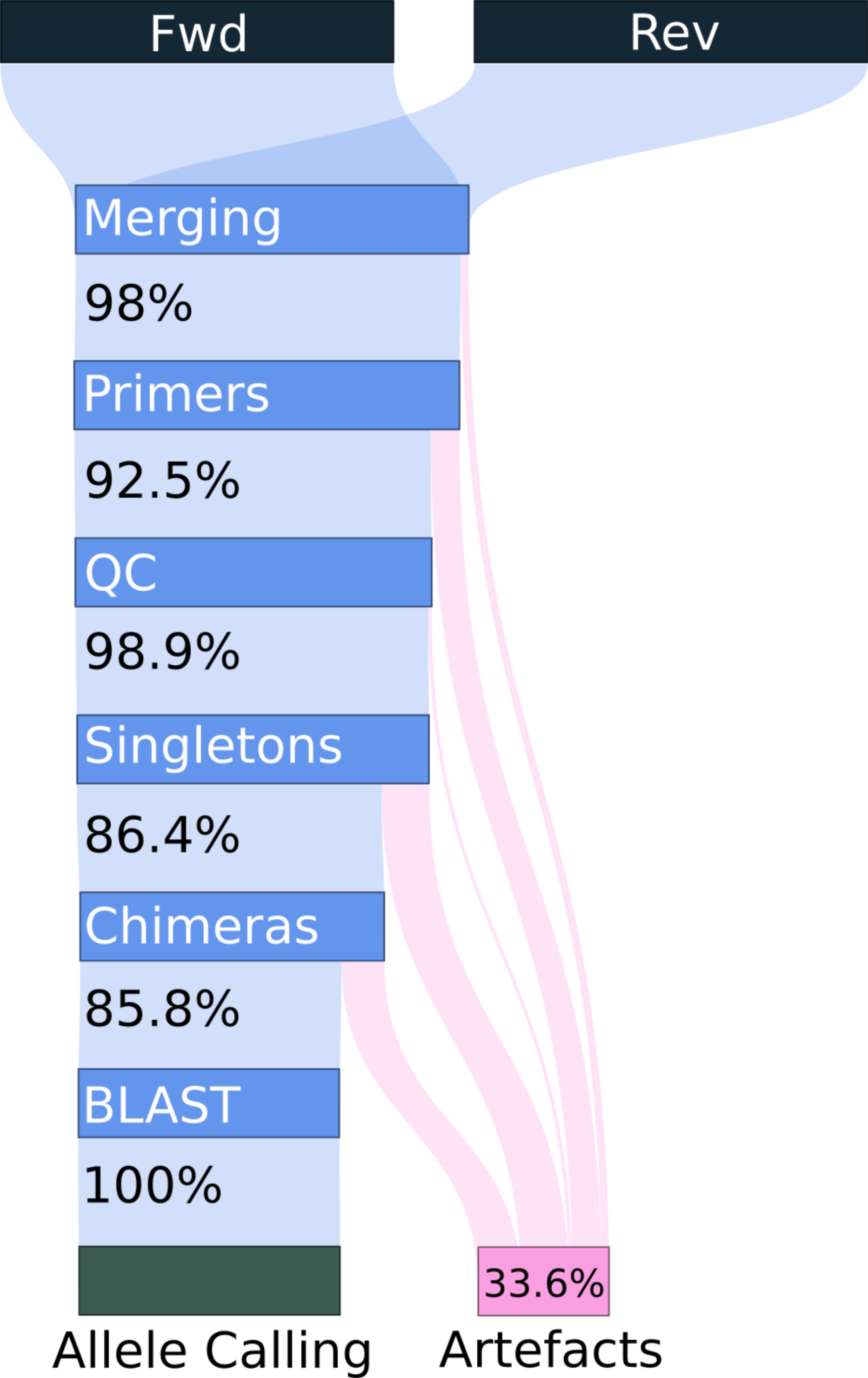
Genotyping novel complex multigene families is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification bias. Most genotyping pipelines have been validated in non-model systems whereby the real genotype is unknown and the generation of artefacts may be highly repeatable. Further hindering accurate genotyping, the relationship between artefacts and genotype complexity (i.e. number of alleles per genotype) within a PCR remains poorly described. Here we investigated the latter by experimentally combining multiple known major histocompatibility complex (MHC) haplotypes of a model organism (chicken, \textit{Gallus gallus}, 43 artificial genotypes with 2-13 alleles per amplicon). In addition to well defined “optimal” primers, we simulated a non-model species situation by designing “cross-species” primers, with sequence data from closely related Galliforme species. We applied a novel open-source genotyping pipeline (ACACIA; \url{https://gitlab.com/psc_santos/ACACIA}), and compared its performance with another, previously published pipeline (AmpliSAS). Allele calling accuracy was higher when using ACACIA (98.5\% vs 97\% and 77.8\% vs 75.2\% for the “optimal” and “cross-species” datasets respectively). Systematic allele dropout of three alleles owing to primer mismatch in the “cross-species” dataset explained high allele calling repeatability (100\% when using ACACIA) despite low accuracy, demonstrating that repeatability can be misleading when evaluating genotyping workflows. Genotype complexity was positively associated with non-chimeric artefacts, chimeric artefacts (nonlinearly by leveling when amplifying more than 4-6 alleles) and allele amplification bias. Our study exemplifies and demonstrates pitfalls researchers should avoid to reliably genotype complex multigene families.