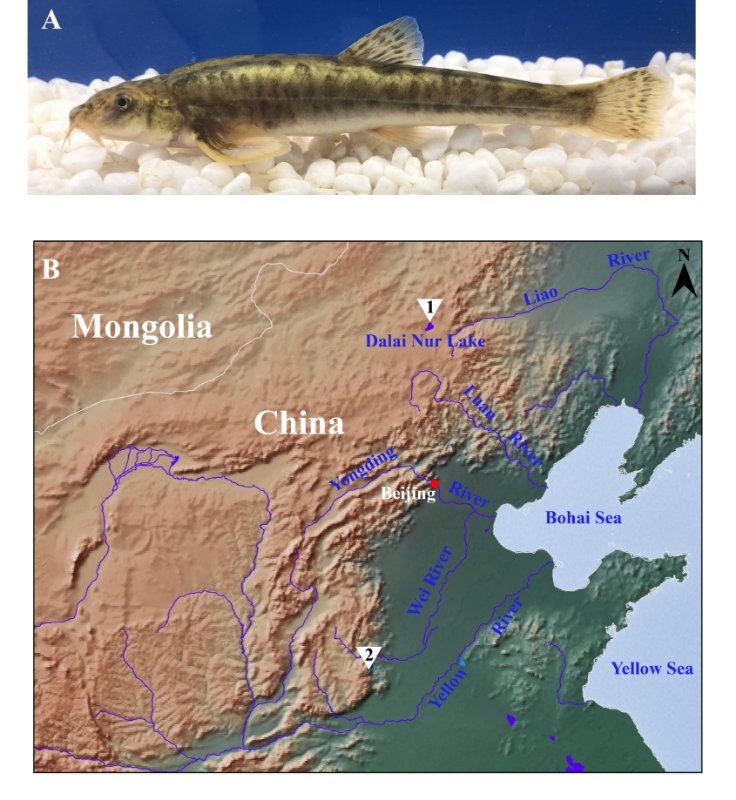
Lake Dali Nur, located in Inner Mongolia, North China, is alkaline, with Triplophysa dalaica one of the three fish species that not only survive, but thrive, in the lake. To investigate the presence of molecular mutations potentially responsible for this adaptation, the whole genome sequence of the species endemic to the lake was sequenced. A total of 126.5 Gb and 106 Gb data, covering nearly 200X of the estimated genome, were generated using long-read sequencing and Hi-C technology, respectively. De novo assembly generated a genome totalled 607.91 Mb, with a contig N50 of 9.27 Mb. Nearly all whole genome sequences were anchored and oriented onto 25 chromosomes, with telomeres for most chromosomes also being recovered. Repeats comprised approximately 35.01% of the whole genome. A total of 23,925 protein-coding genes were predicted, within which, 98.62% could be functionally annotated. Through comparisons of T. dalaica, T. tibetana, and T. siluroides gene models, a total of 898 genes were identified as likely being subjected to positive selection, with several of them potentially associated with alkaline adaptation, such as sodium bicarbonate cotransporter, SLC4A4. Demographic analyses suggested that the Dali population might have diverged from endemic freshwater Hai River populations, approximately 1 million years ago. The high-quality T. dalaica genome, sequenced in this study, not only aids in the analyses of alkaline adaptation, but may also assist in revealing the mysteries of the highly divergent genus Triplophysa.