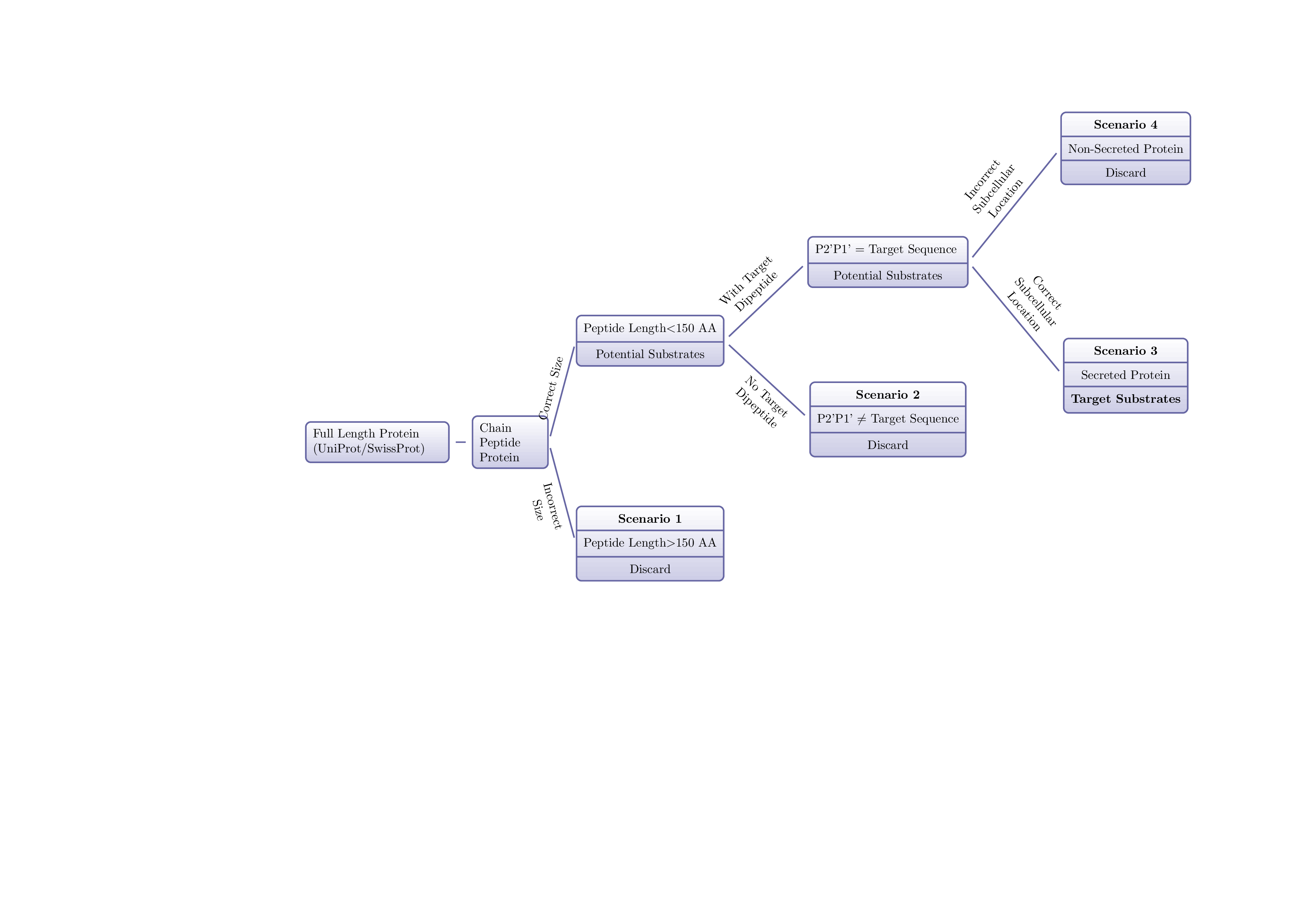
Proteins are arguably one of the most important component in a living organism. As genes encode biological features, proteins deliver biological functions. One protein can adopt many structural conformations, which is largely determined by the minimum overall entropy under that specific sub-cellular environment. Yet, three dimensional conformation and flexibility of a protein determine its functionality by allowing different binding property and hence initiating different cascades events downstream. At molecular level, each protein is composed by several linear poly-peptide chains folding in a specific pattern to give that unique three dimensional shape. Each poly-peptide chain is consisted of a distinct amino acids (a total of 20 types in human) in sequence, and remarkabily, the protein folding sequence and final spatial configuration are largely encoded in the poly-peptide sequences. Unfortunately, the vast scope of possible spatial structural configurations based on a given protein amino acid sequence is almost beyond the exploring capacity (at 10¹⁰⁰⁰ scale) and searching at global scope for the specific structure adaptation with the minimum entropy is extremely computational expensive and time consuming. The rapid development in machine learning recently offers another possibility in rapid determine the protein three s dimensional structure _in silico_ based on the similar protein family. In this study, among the popular unsupervised learning algorithms including Apriori algorithm, k-Means have been trialed, k-Means seems to be the relatively reliable candidate to determine the potential DPP4 substrates from human proteome with 86% accuracy. Further study using Markov Decision Process (MDP) has revealed the peptide/protein backbone torsion angle at serine catalitic site is a major deterministic power in predicting _in vivo_ substrate for DPP4.