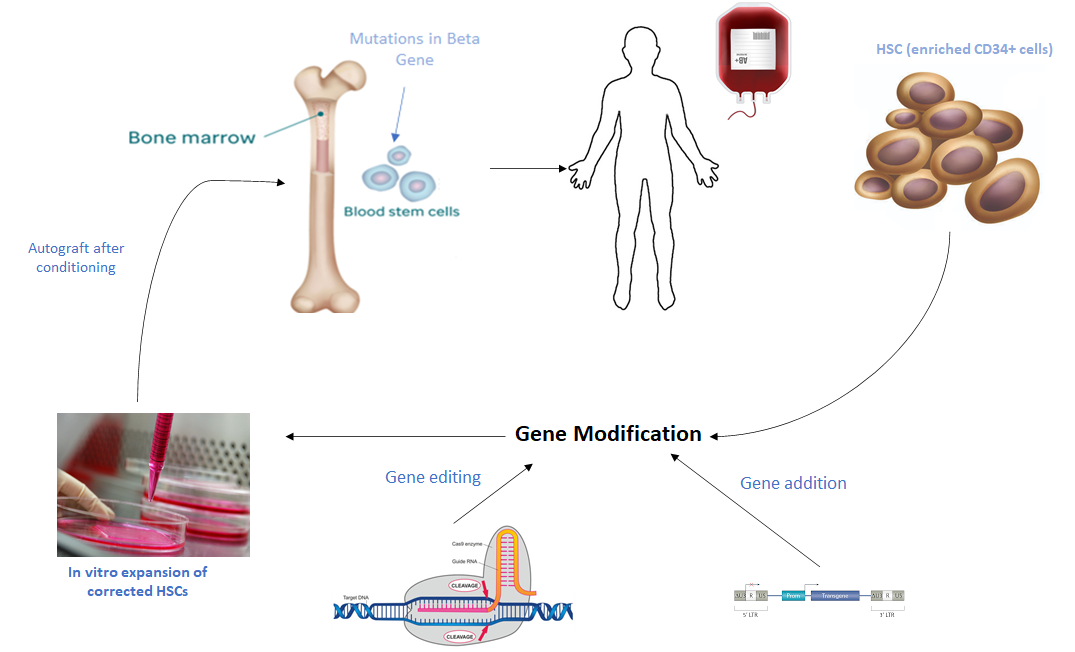
Beta-thalassaemia is one of the most significant haemoglobinopathies worldwide resulting in the synthesis of little or no β-globin chains. Without treatment, β-thalassaemia major is lethal within the first decade of life due to the complex pathophysiology which leads to wide clinical manifestations. Current clinical management for these patients solely relies on repeated transfusions followed by iron chelating therapy which can eventually results into multi-organ damage. A number of novel approaches to correct the resulting α/β globin chain imbalance are currently being developed. These include reactivation of foetal haemoglobin by pharmacological compounds, allogenic hematopoietic stem cell transplantation (HSCT) and gene therapy. Up to now, the only curative treatment for beta-thalassemia is HSCT, but this is a risky and costly procedure. Gene therapy either by gene addition or gene editing is emerging as a powerful approach to treat this disease. Gene addition is currently based on transplantation of autologous hematopoietic stem cells genetically modified with an integrating lentiviral vector expression the globin gene while gene editing involves the use of CRISPR/Cas9 to correct the causative mutation. Although the early outcomes of the clinical trials in gene therapy are showing promising results, they have also highlighted a number of limitations. In this review we will discuss about the current management strategies used to treat beta-thalassaemia and also focus on novel therapies.