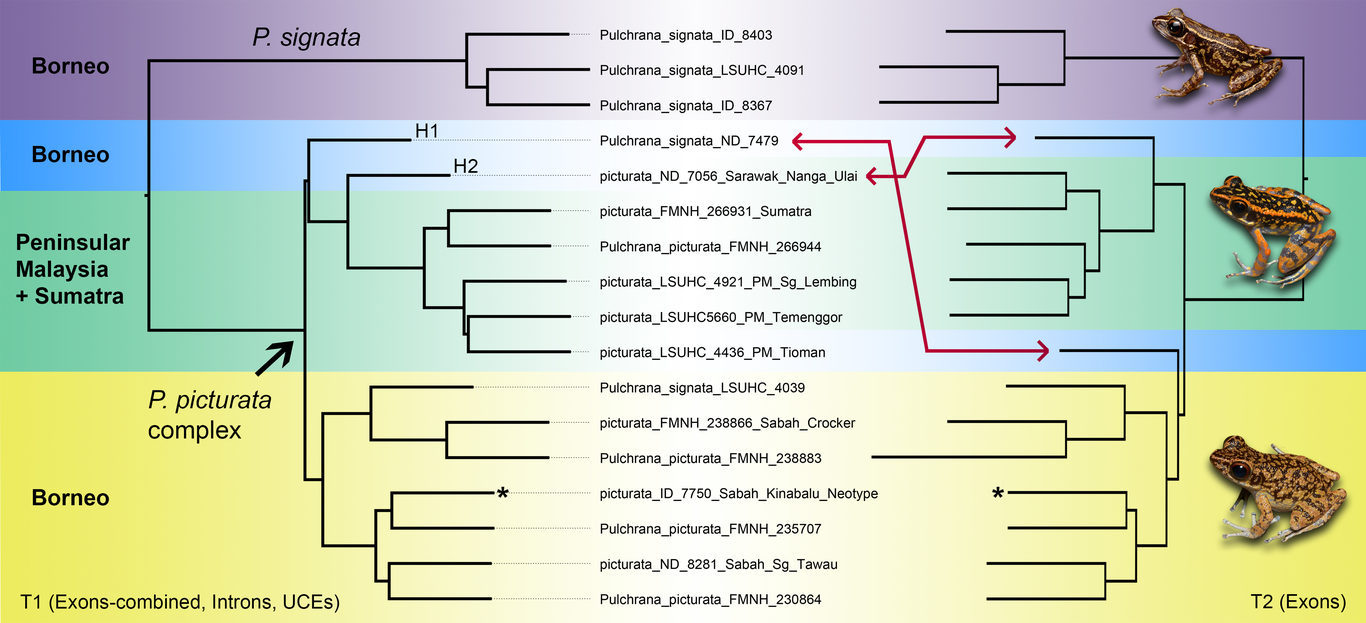
Most new cryptic species are described using conventional tree- and distance-based species delimitation methods (SDMs), which rely on phylogenetic arrangements and measures of genetic divergence. However, although numerous factors such as spatial population structure and gene flow are known to confound phylogenetic and species delimitation inferences, the influence of these processes on species estimation is rarely evaluated. Using large amounts of exons, introns, and ultraconserved elements obtained using the FrogCap sequence-capture protocol, we compared conventional SDMs with more robust genomic analyses that assesses spatial population structure and gene flow to characterize species boundaries in a Southeast Asian frog complex (Pulchrana picturata). Our results showed that gene flow and introgression can produce phylogenetic patterns and levels of divergence that resemble distinct species (up to 10% divergent in mitochondrial DNA). Hybrid populations were inferred as independent (singleton) clades that were highly divergent from adjacent populations (7–10%) and unusually similar (<3%) to allopatric populations. Such anomalous patterns are not uncommon in Southeast Asian amphibians, which brings into question whether the high cryptic diversity observed in other amphibian groups reflect distinct cryptic species—or, instead, highly structured and admixed metapopulation lineages. Our results also provide an alternative explanation to the conundrum of divergent (sometimes non-sister) sympatric lineages―a pattern that has been celebrated as indicative of true cryptic speciation. Based on these findings, we recommend that species delimitation of continuously distributed “cryptic” groups should not rely solely on conventional SDMs but should necessarily examine spatial population structure and gene flow to avoid taxonomic inflation.