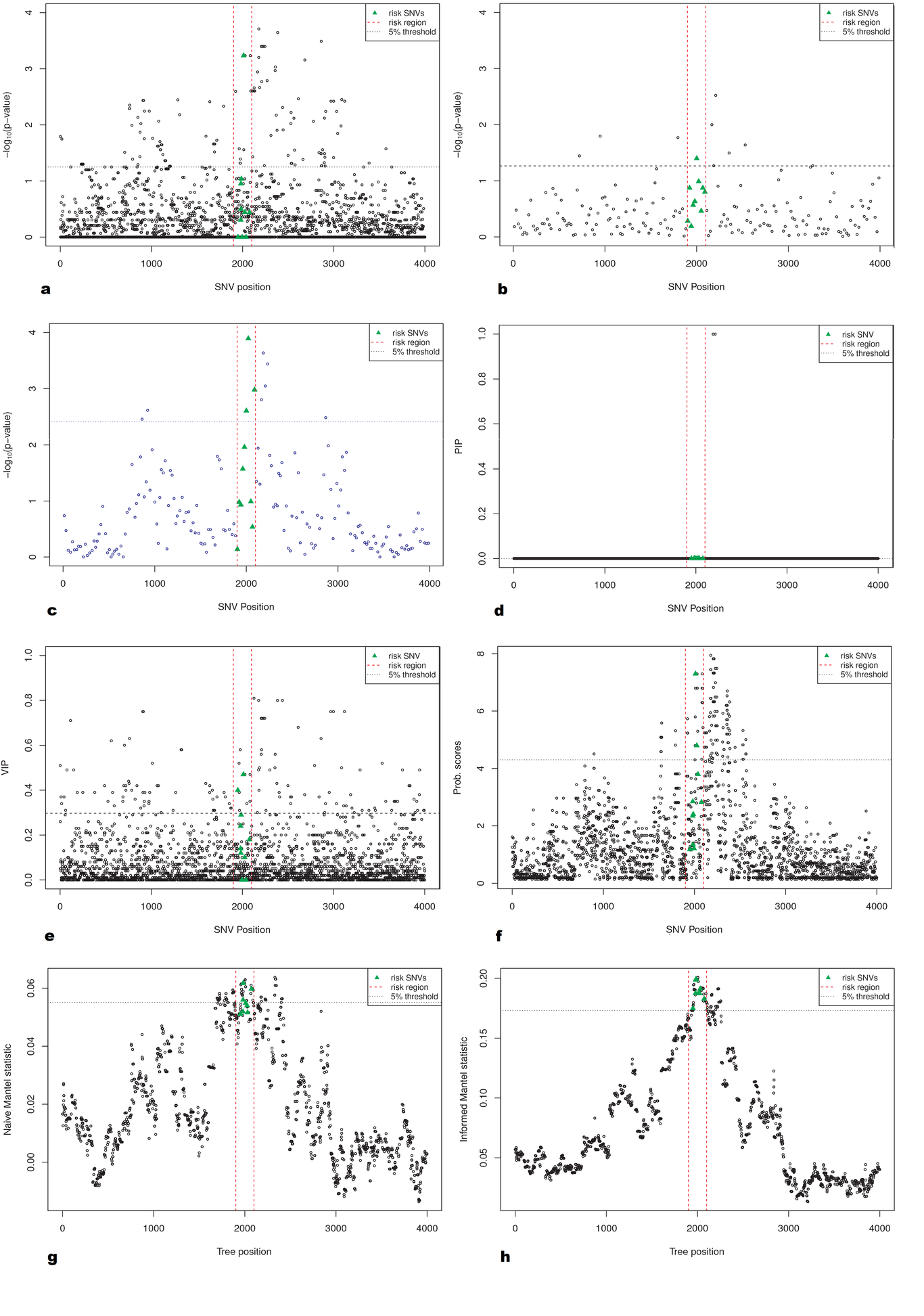
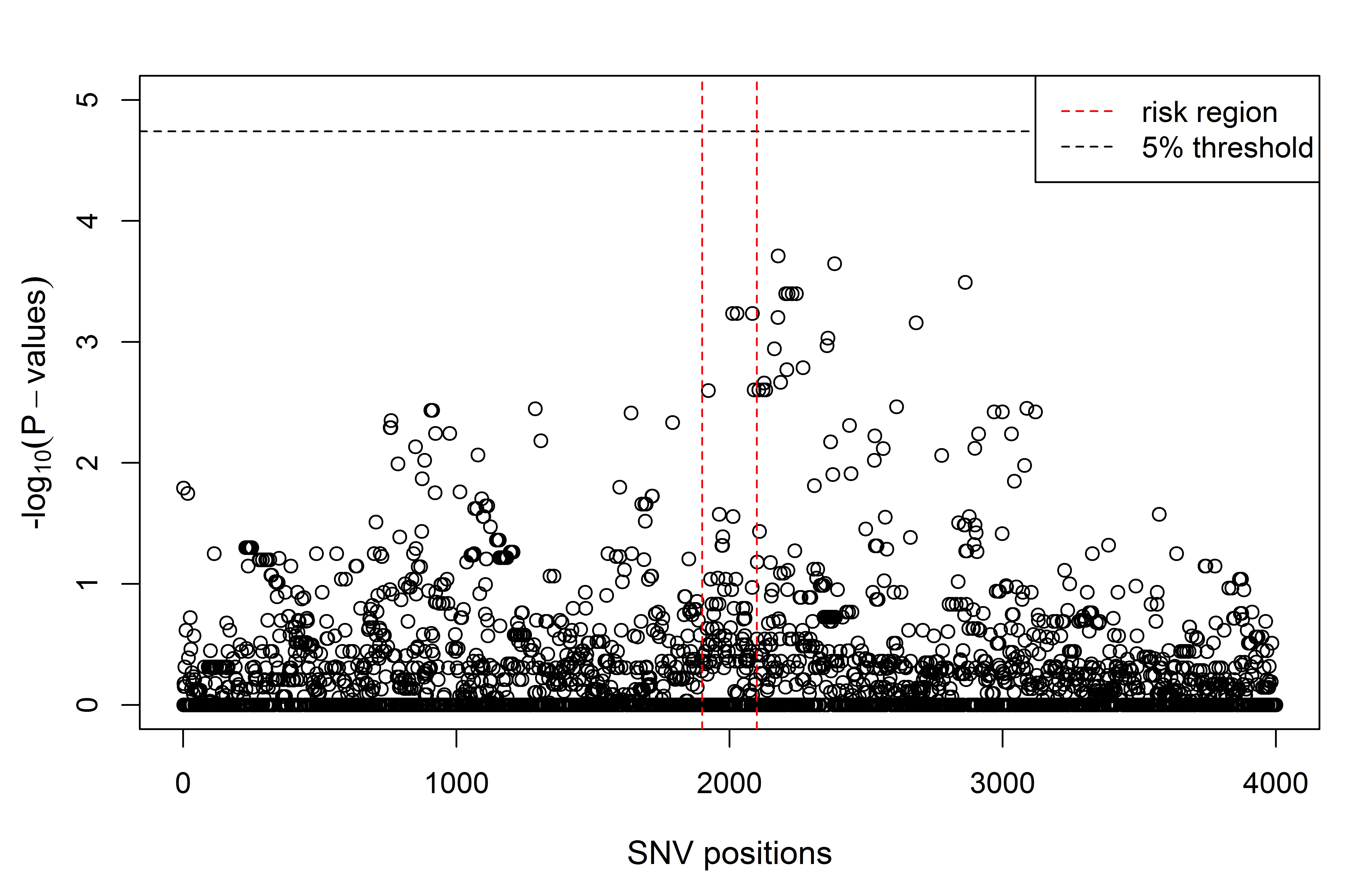
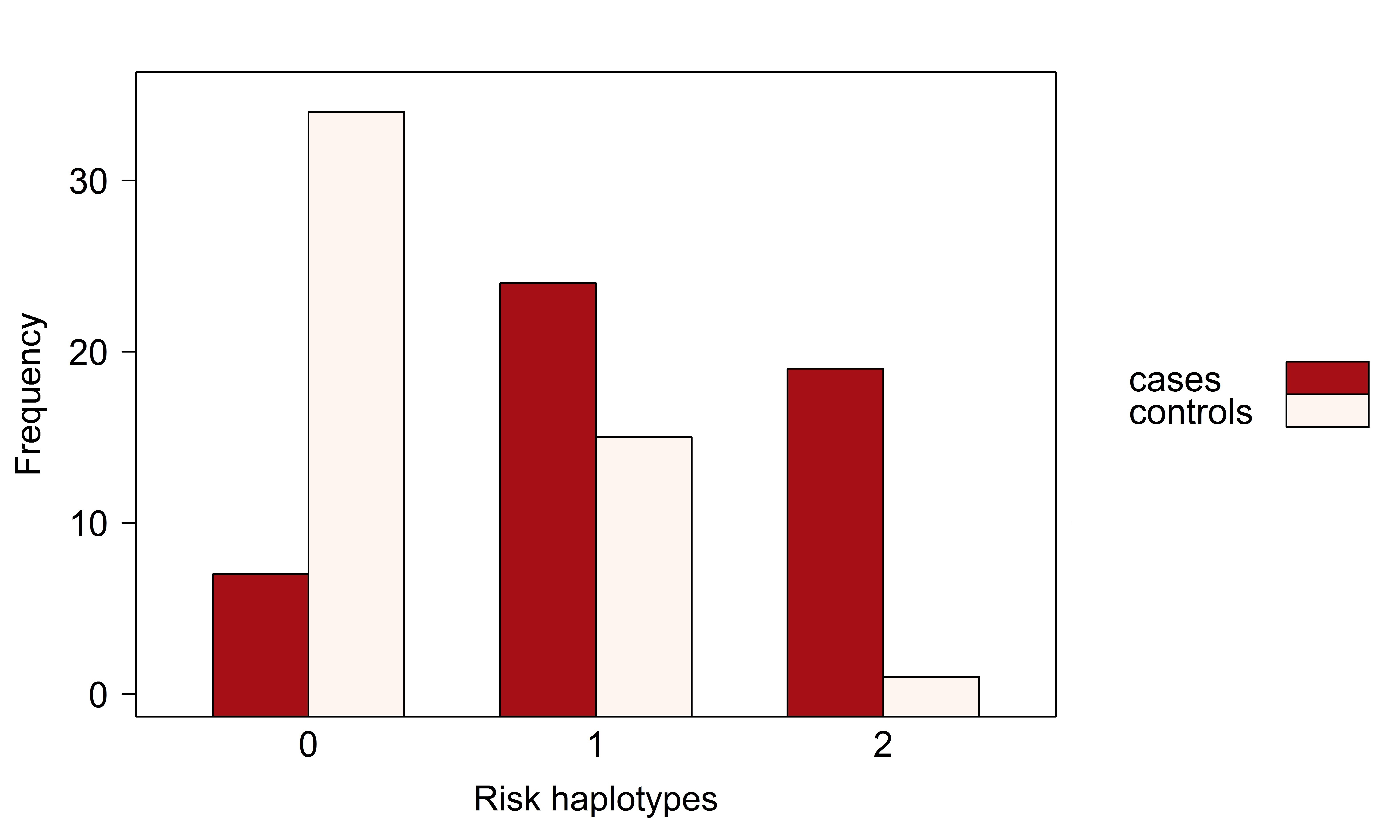
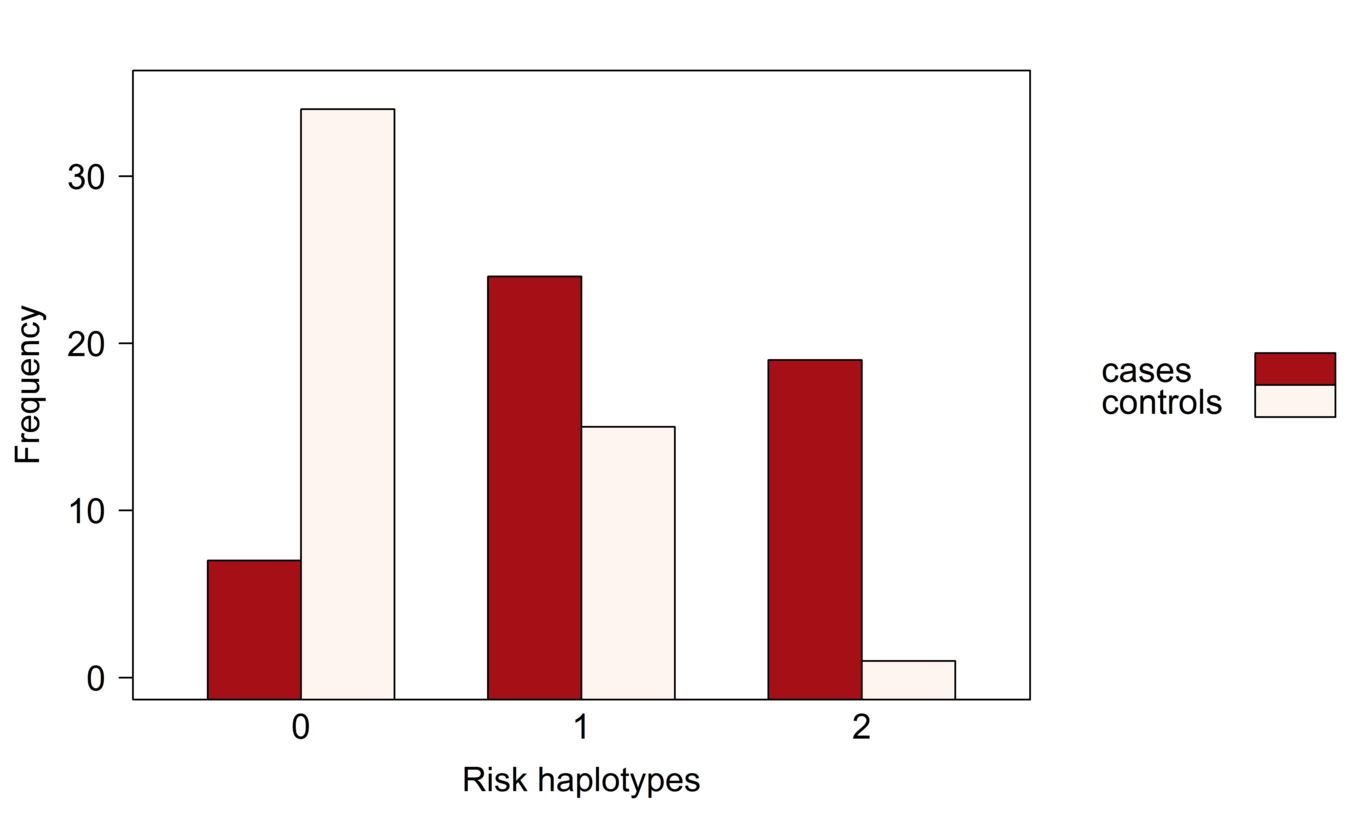
INTRODUCTION Alzheimer’s disease (AD) is a neurodegenerative disorder causing cognitive impairment and memory loss. The estimated heritability of late-onset AD is 60%-80% , and the largest susceptibility allele is the ε4 allele of _APOE_ , which may play a role in 20% to 25% of AD cases. Numerous studies have identified susceptibility genes which account for some of the missing heritability of AD, with many associated variants having been identified through genome-wide association studies (GWAS) \citep[e.g.][]{Beecham_2009, Kamboh_2012, Bertram_2008}. Apart from _APOE_, the associated variants have mostly had moderate or small effect sizes, suggesting that the remaining heritability of AD may be explained by many additional genetic variants of small effect. Identifying susceptibility variants with small effect sizes in GWAS is challenging since strict multiple testing corrections are required to maintain a reasonable family-wise error rate. This analysis focuses on leveraging information from prior family of studies of AD , by looking for association in previously identified linkage regions reported on the Alzgene website . Linkage regions for AD are genomic regions that tend to be co-inherited with AD in families. By definition, linkage regions include susceptibility genes that are co-transmitted with the disease. The regions currently identified from family studies of AD are large, however, since families contain relatively few transmissions. Further transmissions over multiple generations would provide more fine-grain information about the location of susceptibility genes. Previous studies have fine-mapped a single linkage region through association of AD with genetic variants in densely genotyped or sequenced regions , or have confirmed linkage to AD in genomic regions identified from GWAS . In this report, we aim to fine-map multiple linkage regions for AD through multivariate association of their SNPs to the rates of atrophy in brain regions affected by AD. We analyze data from two phases of the Alzheimer’s Disease Neuroimaging Initiative which are case-control studies of AD and mild-cognitive impairment; ADNI-1 and ADNI-2. The rates of atrophy in brain regions affected by AD are so-called endophenotypes: observable traits that reflect disease progression. By investigating the joint association between the genomic variants and the neuroimaging endophenotypes, we use the information about disease progression to supervise the selection of single-nucleotide polymorphisms (SNPs). This multivariate approach to analysis stands in contrast to the commonly-used mass-univariate approach in which separate regressions are fit for each SNP, and the disease outcome is predicted by the minor allele counts. Simultaneous analysis of association is preferred because the reduced residual variation leads to (i) a clearer assessment of the signal from each SNP, (ii) increased power to detect signal, and (iii) a decreased false-positive rate . We also employ inverse probability weighting to account for the biased sampling design of the ADNI-1 and ADNI-2 studies, an aspect of analysis that has not been accounted for in many previous imaging genetics studies . Methods that explicitly account for gene structure have been proposed for analyzing the association between multiple imaging phenotypes and SNPs in candidate genes \citep[e.g.][]{Wang_2011, 1605.02234}. However, these methods become computationally intractable when analyzing data with tens of thousands of genotyped variants. To select SNPs associated with disease progression, we instead use sparse canonical correlation analysis (SCCA) to find a sparse linear combinations of SNPs having maximal correlation with the imaging endophenotypes. Multiple penalty schemes have been proposed to implement the sparse estimation in SCCA . We employ an SCCA implementation that estimates the sparse linear combinations by computing sparse approximations to the left singular vectors of the cross-correlation matrix of the SNP data and the neuroimaging endophenotype data . Sparsity is introduced through soft-thresholding of the coefficient estimates , which has been noted to be similar in implementation to a limiting form of the elastic-net . A drawback of ℓ₁-type penalties is that not all SNPs from an LD block of highly-correlated SNPs that are associated with the outcome will be selected into the model . We prefer an elastic-net-like penalty over alternative implementations with ℓ₁ penalties because it allows selection of all potentially associated SNPs regardless of the linkage-disequilibrium (LD) structure in the data. We may think of SNP genotypes as a matrix X and imaging phenotypes as a matrix Y measured on the same n subjects. \citet*{Robert_1976} showed that estimating the maximum correlation between linear combinations of X and Y in canonical correlation analysis is equivalent to estimating the linear combinations having the maximum RV coefficient, a measure of linear association between the multivariate datasets . As the squared correlation coefficient between the first canonical variates, the RV coefficient is well-suited for testing linear association in our context. We use a permutation test based on the RV coefficient to assess the association between the initial list of SNPs in X and the phenotypes in Y. Although the RV coefficient may overestimate association when n ≪ p , a permutation test with the RV coefficient is preferred over a parametric hypothesis test since the permutation null distribution is computed under the same conditions as the observed RV coefficient, resulting in a valid hypothesis test. The outcome of this test is used to determine whether or not to proceed with a second refinement stage that reduces the number of SNPs by applying SCCA. Selection of the soft-thresholding parameter in SCCA is challenging in our context. Since the number of SNPs exceeds the sample size and many of the SNPs are expected to be unassociated with the phenotypes, large sample correlations can arise by chance . Indeed, the prescribed procedure of selecting the penalty parameter with highest predicted correlation across cross-validation test sets results in more than 98% of the SNPs remaining in the model. A prediction criterion for choosing the penalty term may contribute to the lack of variable selection, allowing redundant variables into the model . When the same tuning parameter is used for variable selection and shrinkage, redundant variables tend to be selected to compensate for overshrinkage of coefficient estimates and losses in predictive ability . In our case, there is effectively no variable selection and little insight is gained by allowing for sparsity in the solution. To circumvent the lack of variable selection from SCCA, we fix the tuning parameter to select about 10% of the SNPs and then use resampling to determine the relative importance of each SNP to the association with neuroimaging endophenotypes. Instead of using the prediction-optimal penalty term, we fixed the soft-thresholding parameter for the SNPs to achieve variable selection based on the rationale that no more than about 7,500 SNPs, or approximately 10%, are expected to be associated with the phenotypes. This choice is guided by prior experience in genetic association studies, where the majority of genetic variants have no effect on the phenotypes, or an effect that is indistinguishable from zero . The organization of the manuscript is as follows. The Materials and Methods section describes the ADNI data, the data processing procedures, and the methods applied for discovery, refinement, and validation. The Results section presents the results of the analyses. The Discussion section notes challenges and successes of the analysis, including considerations for modelling continuous phenotype data under a case-control sampling design, and provides interpretation of the results.